Sequence Ontology FAQs Index
Ontology Questions
- What is the Sequence Ontology?
- How is SO different to GO?
- What are the advantages of using an ontology over a controlled vocabulary to describe sequence data?
- How is so different to the existing sequence feature table used by the main databanks?
- What are relationships?
- What are properties?
- What is transitivity?(or why is it that mRNA sequence cannot contain introns?)
- How can I view SO?
- How can I contribute to SO?
- Who can contribute to or use SO?
- How should I suggest a new term to SO?
- How do I become a contributor to the wiki?
- How do I cite SO?
- Do I need a licensing agreement to use SO?
- How do I download the ontology?
Format Questions
- What is OBO format?
- How do I parse OBO files?
- Where is the schema?
- Which databases and file formats use SO?
Annotation Questions
- How should I use SO to annotate my new sequence?
- Can I convert existing annotations into SO compliant annotations?
- Do other controlled vocabularies map to SO?
- What is the difference between SO and SOFA?
Analysis Questions
Ontology Questions
What is the Sequence Ontology?
The Sequence Ontology (SO) project aims to unify the way in which we describe sequence annotations, by providing a controlled vocabulary of terms and the relationships between them. The terms in the ontology describe the features and properties of biological sequence. Features occupy a length of sequence such as ‘exon’, whereas properties describe attributes of the features. For example a gene may have the property ‘maternally_expressed’. Alone the phrase ‘maternally_expressed’ cannot be located onto a sequence, but must be associated with a feature.
The concepts in the ontology are structure by relationships, which allow classification hierarchies to be built. Each relationship has a defined meaning, which allows the connection between the two concepts to be understood. The position of a concept in relation to others also provides some context to the meaning of the concept. The defined relationships between the features are: subclass [is_a], membership [part_of], topological [adjacent_to] and derivation [derives_from].
We use the term ‘ontology’ to mean a representation of our ideas about a given domain, with the intention that these ideas can be processed and used both computationally and manually. In his guest editorial for AI Topics, Welty provides a summary of ‘what is an ontology’ with regards to computer science.
How is SO different to GO?
The Gene Ontology is a collection of terms used to describe gene products. It is divided into three aspects, cellular location, biological process and molecular function. A gene product may be described by one or more terms from each of the aspects. The characterization of gene products in this way enhances many large scale analyses. The Sequence Ontology however, is used to catalog the features and properties of biological sequence, and how these features relate to each other. It captures the subclass hierarchy of these features, the meronomies, that is what features are parts of other features, and also topological relationships like adjacent_to. Using SO and GO together to catalogue the kinds of gene products and the features of the sequence will mean that queries such as “Give me the 3’UTR sequence of all transcription factors in the X pathway” will be trivial.
What are the advantages of using an ontology vs. a controlled vocabulary to describe sequence data?
A controlled vocabulary is a list of terms and sometimes synonyms used to describe the concepts. An ontology not only provides a unified vocabulary of terms to describe the concepts but also relates the concepts together into hierarchies and networks, that provide the basis for inference and automated reasoning.
Some advantages of using SO over a controlled vocabulary are:
- Specifying the relationships between the features means that SO-compliant software need only be aware of the defined relationships and be provided with an up to date ontology. Software need not hard code the facts since that knowledge is in the ontology; it need only be able to navigate the relationships. When a new term is added to the ontology, the software will know what kind of "thing" it is, and what relationships and properties it can have.
- The added depth of knowledge in the ontology improves the searching and querying capabilities of software that utilizes it. An analysis that requires the non-coding RNA sequences need only query for features that are kinds of (is_a) non_coding_RNA, rather than specify each type such as tRNA, miRNA etc.
How is so different to the existing sequence feature table used by the main databanks?
The Feature Table is a controlled vocabulary of terms describing sequence features and is used to describe the annotations distributed by the large genome databanks (GenBank, EMBL and DDBJ). It does provide a grouping of its terms for annotation purposes, based on the degree of specificity of the term. For example, CAAT_signal is more specific than promoter and both 5’UTR and mRNA are more specific than precursor_RNA. These relationships between the terms are not formalized, thus the interpretation of them is left to the user to infer, and, more critically, must be hard-coded into software applications. This is in contrast to SO where these relationships are explicitly defined. 5_prime_UTR is part_of a transcript, and mRNA is_a kind of transcript.
The terms describing sequence features in SO and SOFA are richer than those of the Feature Table. Most of the terms in the Feature Table map directly to terms in SO, although the term names may have been changed to fit the SO naming conventions. In general, SO contains a more extensive set of features for detailed annotation. At the time of writing there are 171 locatable sequence features in SOFA compared to 65 of the Feature Table. There are 11 terms in the Feature Table that are not included in SO. These terms fall into two categories: remarks and immunological features, both of which have been handled slightly differently in SO.
When annotating features with terms in SO, attempt to specify the deepest node in the tree possible for that feature, with the available information. For example, mRNA conveys more information than transcript. As there is a subclass hierarchy, terms closer to the root are more general. SO, unlike the feature table does not contain catch-all miscellaneous terms such as misc_feature, misc_signal or misc_RNA. There are more general terms within the ontology that capture these less specified concepts ( sequence_feature, regulatory_region and transcript respectively).
What are relationships?
Relationships are the logical connections between concepts; the arcs between the nodes. OBO has proposed ten relationships and definitions in the paper Relations in Biological Ontologies by Smith et al 2005. Providing we have defined the meaning of the relationships, traversing the concepts in the ontology via the relationships allows us to understand more about what a concept is. If we follow the is_a relationships of a node back to the root, we will gain some understanding of what the concept is, likewise if we follow the part_of relationships we will be able to place the concept into a composite whole. Traversing the concepts in this way is the basis of inference and reasoning.
What are properties?
In the same way that a rose may have the property of being red, features in SO may also have properties. Properties are the attributes of a feature that further describe it, but cannot exist alone without the feature. Red does not exist except to describe another medium such as paint, the sky and flowers. In SO there is an aspect of the ontology that details the properties of sequence_feature, called sequence_attribute.
The sequence attributes provide more information about the specific kinds of feature. There are transcript_attributes such as mRNA_with_frameshift, transpliced_transcript and multicistronic_transcript. There are attributes that refer to the gene such as imprinted and post_translationally_regulated. There are attributes that refer to the results of experiments such as CDS_predicticted versus CDS_independently_known.
There is an aspect of the ontology, consequences_of_mutation, that deals with the impact that the mutation has, be it affecting splicing, the regulatory region etc. There is also an aspect of the ontology, chromosome_variation, that details the types of chromosome mutation with terms like aneuploid and transposition.
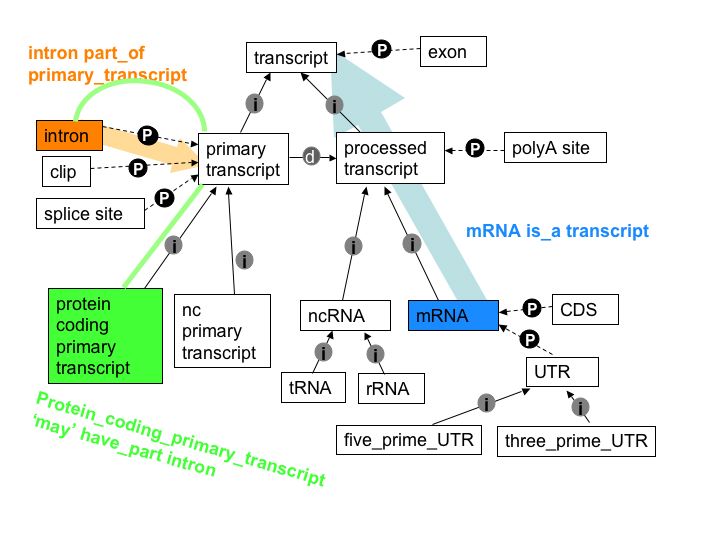
What is transitivity? (Or why is it that mRNA sequence cannot contain introns?)
Transitivity is the process of traversing between concepts using the relationships and being able to infer conclusions from this. Each relationship allows us to infer different information. The is_a relationship allows us to infer what something is. For example, if we think about the is_a relationships of the Linnaean taxonomy, we can see that human is_a primate, and primate is_a mammal, and thus infer that human is_a mammal.
This inference can come in useful when we want to learn about the properties of a concept. Through the is_a relationship, a sub-type concept (hyponym) inherits the properties and relationships of its super-type concept (hypernym). This is because the high-level concept groups together more specialized concepts that share things in common. To go back to the Linnaean example, a property of mammals is they feed their young milk, therefore primates also inherit this property.
In the Sequence Ontology transitivity can be used to validate the contents of an annotation against what is known in the ontology. The following relationships are stated in the ontology:
- primary_transcript is_a transcript
- processed_transcript is_a transcript
- intron is part_of a primary_transcript
- mRNA is_a processed_transcript
- protein_coding_primary_transcript isa primary_transcript
If an annotation suggested that an intron was part of an mRNA the relationships between the concepts could be traversed to see if this is possible.
An mRNA is_a transcript (transitively via processed trancript). An intron however is part_of a primary_transcript, thus subtypes of primary_transcript may contain introns. This means that it is valid for a protein_coding_primary_transcript to have an intron as a part. There is no relationship link between mRNA and intron that would allow an intron to be part of mRNA.
How can I view SO?
SO can be viewed online with the miSO onotology browser. This browswer allows searching by term, ID or synonym. Each term in the ontology browser links to the corresponding page in the SO wiki where community documentation of terms is encouraged.
SO is archived in OBO format, and is best viewed using the OBO-Edit ontology editor. OBO-Edit is an ontology editor, developed by the Gene Ontology Consortium. It allows both browsing and editing of the structure and contents of the ontology, see Figure 1. The ontology is maintained in the SO CVS archive.
For quick reference, the SO terms are also documented in an HTML navigable flat file, which shows the definitions, and relationships between terms.
How can I contribute to SO?
The main portal for community documentation of the Sequence Ontology is the SO wiki. Here is the place to add annotation and documentation of SO terms. Discussion about SO and comments pertaining to the extension of SO are addressed via the SO-developers mailing list. Anyone can join this list or post a message. The archives of previous discussions are also available.
Who can contribute to or use SO?
SO is part of OBO and is therefore subject to the rules of OBO, namely “The ontologies must be open and can be used by all without any constraint other than that their origin must be acknowledged and they cannot be altered and redistributed under the same name.” The target audience of SO is those in the model organism community, annotating whole genomes. It is hoped that SO will provide a unified language for describing annotations. All interested parties are encourages to contribute annotation and documentation for SO terms on the SO wiki and to participate in disucssion of all things SO on the SO-developers mailing list.
A mailing list exists which acts as a forum for discussion of the ontology and related issues. Anyone can post to, or join this list. The archives of previous discussions are also available.
How should I suggest a new term to SO?
The best way to suggest a new term to SO is to use the term tracker. Your query will be logged and we will get back to you via the tracker for further clarification. It is good practice to give your entry a sensible name such as the term you are suggesting, and also a definition and links to references about your new term. If you are not anonymous, it is easier to continue a discussion with you.
How do I contribute to the SO wiki?
You must become a registered user to contribute to the SO wiki. Any registered user can set up a new account for a colleague. Or just send us an email keilbeck@genetics.utah.edu to tell us who you are, and we'll set up an account for you. Be sure to send us your preferred login name.
How do I cite SO?
Please cite this paper:
The Sequence Ontology: A tool for the unification of genome annotations. Eilbeck K., Lewis S.E., Mungall C.J., Yandell M., Stein L., Durbin R., Ashburner M. Genome Biology (2005) 6:R44
Please also include the version of SO or SOFA used, or the SOFA release number if applicable.
Do I need a licensing agreement to use SO?
The Sequence Ontology is in the public domain. It is available to public and private sector users with no licensing requirements. We ask that you cite SO and do not make changes to SO then redistribute under the same name.
How do I download the ontology?
SO can be downloaded via CVS, either via anonymous CVS or by browsing the CVS tree.
Format Questions
What is OBO format?
The Sequence Ontology is developed and archived using OBO format. OBO stands for Open Biomedical Ontologies. It is a collection of structured controlled vocabularies for shared use across the biomedical domain, such as Gene Ontology. A file format for sharing ontologies has been developed by the OBO group.
How do I parse OBO files?
A suite of Perl modules called go-perl is available from CPAN.
For Java people, OBO-edit includes a modular library for parsing and serializing OBO files. Users can either parse OBO files into the OBO-Edit data-model or they can plug in their own event based parser. The toolkits are available from the Gene Ontology CVS repository. Users are strongly advised to develop their software using the OBO-Edit libraries, as they are being actively developed. The documentation however is still small, and mostly contained within the source code comments.
Where is the schema?
Being an ontology, SO transcends any particular database schema or file-format. This means it can be used equally well as an external data exchange format or internally as an integral component of a database. The CHADO database from the GMOD community uses SO to type its features. There is not a single specific Sequence Ontology database.
Which databases and file formats use SO?
SO and SOFA are especially well suited for use with the Generic Feature Format. The latest version, GFF3 uses SOFA terms to standardize the feature type described in each row of a file and SO terms as optional attributes to a feature.
SO can also be employed within a database. CHADO is a modular relational database schema for integrating molecular and genetic data and is part of the Generic Model Organism Database project (GMOD). The CHADO relational schema is extremely flexible, and is centered on genomic features and their relationships, both of which are described using SO terms. This use of SO ensures that software that queries, populates, and exports data from different CHADO databases is interoperable, and thus greatly facilitates large-scale comparisons of even very complex genomics data.
Chaos-XML is a file format that uses SO to label and structure data; but it is intimately tied to the CHADO. Chaos-XML is a hierarchical XML mapping of the CHADO relational schema. Annotations are represented as an ontology-typed feature-graph. The central concept of Chaos-XML is the sequence-feature, which is any sequence entity typed by SO. The features are interconnected via feature relationship elements, whereby each relationship connects a subject feature and an object feature. Features are located via featureloc elements which use interbase coordinates. Chaos-XML and CHADO are richer models than GFF3 in that feature_relationships are typed, and a more sophisticated location model is used. Chaos-XML is the substrate of a suite of programs called Comparative Genomics Library (CGL).
Annotation Questions
How should I use SO to annotate my new sequence?
The paper: Sequence Ontology Annotation Guide. Karen Eilbeck and Suzanna E. Lewis. Comparative and Functional Genomics (2004) 5:642-647 provides an outline to creating a SO compliant annotation, using GFF3 as the file format.
At a minimum, the features of the sequence should be labeled with the most specific term applicable. It is also possible to annotate sequence as you would for GenBank, and then use one of the available scripts to convert to SO.
Can I convert existing annotations into SO compliant annotations?
There are a variety of scripts, software libraries and web applications available to convert existing annotations into SO compliant Chaos XML or GFF3 annotation formats.
- The Chaos-XML Library provides several scripts to convert a variety of file formats into a SO compliant Chaos-xml file.
- The BioPerl project provides a script genbank2gff3.PLS which will convert NCBI GenBank formatted files into SO compliant GFF3 format. As well as a variety of other perl scripts and libraries for working with GFF formatted annotations.
- There is a web based GTF to GFF3 Converter as part of this web site. It will convert GTF (such as TwinScan output) formatted files into valid GFF3. The perl script gtf2gff3 that runs this application is also available for download and can be run as an indepent command line tool.
Do other controlled vocabularies map to SO?
A mapping is being developed between the Feature Table of the genome databanks and SO.
A mapping has been developed by MGED ontology where they will use SO terms for the sequence concepts.
What is the difference between SO and SOFA?
SOFA stands for Sequence Ontology Feature Annotation, and aims to capture a subset of the terms in SO that can be located on a sequence. The purpose of this subset is similar in principle to the idea of GO-slims. It provides a smaller more stable set of terms for use primarily in automated or semi-automated annotation pipelines. SOFA does not contain any terms that are properties.
Analysis Questions
Are there any code libraries that can manipulate SO annotations?
The Comparative Genomics Library (CGL) is a Perl Library that uses SO-compliant chaos-xml annotations as input.
Bioperl includes a module Bio::FeatureIO::gff.pm which ‘reads a feature record from a GFF stream and returns it as an object’.